ABSTRACT
Carbon capture and storage is a vital strategy for mitigating rising atmospheric carbon dioxide, and metal–organic frameworks (MOFs) have gained attention as promising sorbents. Numerous simulations have examined factors governing CO2 capture in MOFs—such as diffusion in MOF-74 under varying temperatures and process modeling of MOF-5—but most were limited to specific structures or conditions, hindering a systematic understanding of diffusion across diverse MOFs. Conventional computational methods also face constraints: density functional theory mainly provides static energy evaluations, while molecular dynamics relies on fixed force fields with poor transferability and an inability to describe reactive events. To overcome these limitations, this study employs molecular dynamics simulations driven by neural network potentials to evaluate CO2 diffusivity in 17 types of MOFs. Results reveal significant variation in transport behavior, with zeolitic-imidazolate framework-3 showing the highest diffusivity and MOF-74 the lowest—an approximately 19-fold difference. These findings highlight the capability of neural-network-based molecular dynamics to deliver consistent and quantitative assessments of CO2 transport in MOFs, providing a reliable framework for the rational design of next-generation capture materials.
-
KEYWORDS: Metal-organic framework, Neural network potential, Molecular dynamics simulation
-
KEYWORDS: 금속-유기 골격체, 신경망포텐셜, 분자동역학 시뮬레이션
1. 서론
대기 중 이산화탄소 농도는 육상 생물권과 해양 사이의 활발한 교환에 의해 계절적으로 변동하며, 광합성·호흡과 해양의 흡수 및 방출 과정이 주요 요인으로 작용한다. 20세기 이후 대기 중 이산화탄소 농도는 화석연료의 연소와 산업화의 영향으로 꾸준히 증가하여, 산업화 이전 약 280 ppm에서 2000년도에는 380 ppm에 도달하였다[
1]. 이는 약 30% 이상의 상승으로, 지구 시스템의 에너지 불균형과 온실효과 강화를 초래하고 있다. 이러한 대기 중 이산화탄소의 증가는 지구 온실효과를 더욱 강화시켜 평균 기온 상승을 촉진하며, 해빙과 빙하의 급격한 감소로 이어져 해수면 상승을 유발한다[
2,
3]. 그 결과 저지대 및 섬 국가의 침수 위험이 높아지고, 극한 기후현상의 빈도와 강도 또한 증가한다. 이로 인해 강우 패턴의 변화, 가뭄과 홍수의 확대가 발생하며, 농업 생산성 저하, 물 자원 부족, 생태계 붕괴 및 사회 기반시설 피해가 심화되고 있다. 이러한 기후·생태·사회 시스템 전반의 변동성 증가는 인류 사회가 직면한 지속가능성 위기를 더욱 복합적이고 심각하게 만든다. 이러한 문제를 해결하기 위해서는 탄소 포집·저장(Carbon Capture and Storage, CCS) 기술이 필수적인 전략으로 간주되며 특히 화학 플랜트와 같은 대규모 배출원의 연소 가스로부터 이산화탄소를 분리·회수하는 포집 기술이 핵심적이다[
4-
6]. 현재 다양한 형태의 포집 소재가 연구되고 있으며, 대표적으로 액상 흡수제(Liquid Absorbent), 금속-유기 골격체(Metal-organic framework, MOF) 기반 고상 흡착제, 분리막(Membrane) 전기화학적 장치가 있다. 액상 흡수제는 주로 아민계 물질로 구성되며, 높은 선택성과 운전 안정성으로 널리 활용되어 왔으나, 높은 탈착 에너지 요구, 아민의 열화 및 부식 문제라는 한계를 가진다. 이를 극복하기 위한 대안으로, 낮은 탈착 에너지, 높은 흡착 용량, 우수한 선택성, 그리고 구조 설계 유연성을 특징으로 하는 MOF 기반 흡착제 연구가 활발히 진행되고 있다[
7-
10].
MOF는 금속 이온 또는 금속 클러스터가 유기 리간드와 배위 결합하여 형성되는 다공성 결정성 소재로, 금속의 종류와 유기 리간드에 도입된 기능기에 따라 결정 구조와 기공 특성을 정밀하게 제어할 수 있다. 이러한 구조적 설계의 유연성은 가스 저장·분리, 액상 정제, 에너지 저장, 촉매 등 다양한 응용 분야에서 성능 향상을 가능케 하는 핵심 요인으로 작용한다[
4,
5]. 지난 수십 년간 MOF의 합성 및 응용에 관한 연구는 비약적으로 발전해 왔으며, 현재도 활발히 진행되고 있다. 특히 이산화탄소 포집 과정에서 MOF의 개방 금속 자리는 강력한 흡착 지점으로 작용하는 것으로 알려져 있으며, 금속의 종류와 기공 구조의 차이는 확산 거동과 체류 시간에 직접적인 영향을 미친다[
9-
12]. 따라서 MOF 내 이산화탄소 확산 성능을 정량적으로 평가하는 것은 실제 적용을 위한 필수적인 단계이다. 현재까지 보고된 약 20,000여 종의 MOF를 모두 실험적으로 검증하는 것은 막대한 시간과 비용이 소요되므로, 전산모사를 이용한 사전 스크리닝이 필요하다. 대표적으로는 분자동역학(Molecular Dynamics, MD) 시뮬레이션을 활용하여 입자거동을 모사할 수 있지만, 이 기법이 적용 가능한 금속 원자 개수는 제한되는 등 범용적으로 이용하기엔 제약이 따른다[
13,
14]. 최근 신경망 포텐셜(Neural Network Potential, NNP)을 적용한 MD 시뮬레이션이 등장하면서 금속-산화물의 상호작용을 보다 정확하게 모사할 수 있는 가능성이 제시되고 있다. 본 연구에서는 17종의 MOF를 선정하여 주요 금속 종류에 따른 이산화탄소의 내부 확산 거동을 분석하였다. 모든 시스템은 NNP 기반 MD 시뮬레이션을 기반으로 수행되었으며, 이로부터 각 MOF 내 확산계수를 도출하였다. 그 결과, Zeolitic Imidazolate Framework-3 (ZIF-3)은 7.63 × 10
-19 cm
2/s로 가장 높은 확산계수를 보였으며, MOF-74는 4.01 × 10
-20 cm
2/s 가장 낮은 값을 나타내어 약 19배의 차이를 보였다. 이는 금속 원소의 종류에 제한되지 않으므로, NNP 기반 MD 시뮬레이션으로부터 MOF 내 이산화탄소의 확산 거동을 정량적으로 평가하는 데에 적합한 기법인 것으로 판단된다.
2. 방법론
2.1 시뮬레이션 모델 구성 및 설정
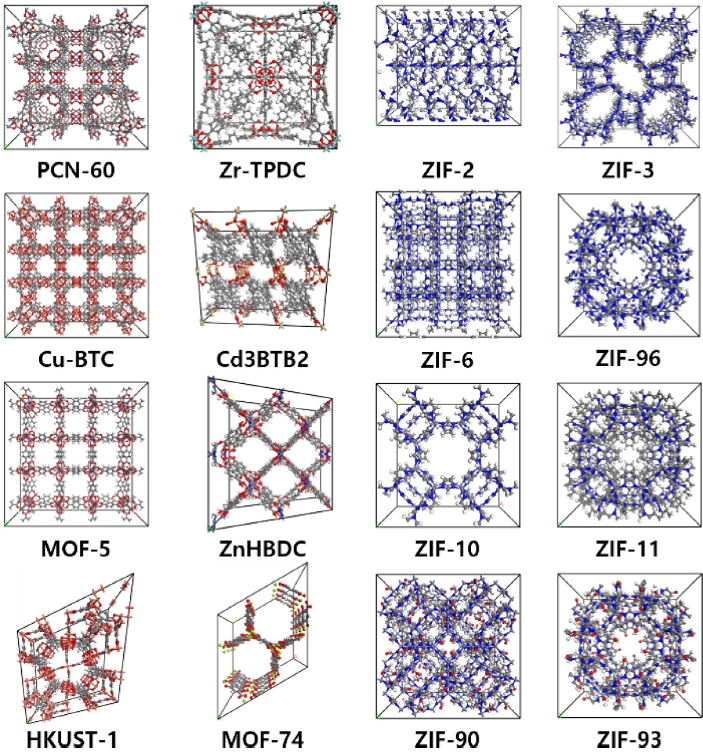
MOF 내 이산화탄소의 확산 거동을 모사하기 위하여 전이금속 종류에 따라 구분된 17종의 MOF를 선정하였다. 대표적인 전이금속으로는 Zn, Cu, Co가 있으며, 이들은 다양한 배위 환경을 형성할 수 있어 MOF의 구조적 안정성과 흡착 특성에 결정적인 영향을 미치는 것으로 알려져 있다. 이러한 금속적 특성을 고려하여 전이금속 기반 MOF를 분석 대상으로 선정하였으며, 본 연구에서 확산도 평가에 활용된 일부 MOF 유닛 셀 구조를
Fig. 2에 제시하였다. 특히 이산화탄소 포집 성능은 흡착 용량 뿐만 아니라 기공 내부의 확산 특성과 밀접하게 연관되므로[
15], 확산계수를 핵심 지표로 설정하였다.
MD 시뮬레이션은 Preferred Potential (PFP)라 불리는 보편적 신경망 포텐셜(Universal Neural Network Potential, UNNP)을 기반으로 수행되었다. 본 연구에서 도입한 보편적 신경망 포텐셜의 일종인 PFP는 밀도범함수이론(Density Functional Theory, DFT)의 데이터 셋을 기반으로 학습된 인공 신경망 포텐셜이다[
16].
시뮬레이션 시스템은 이산화탄소와 질소 분자를 1 : 1 비율로 포함하도록 구성하였으며, 총 밀도가 1.8 g/cm3가 되도록 설정하였다. MOF 구조는 2 × 2 × 2로 확장한 단위셀을 적용하였으며, 이에 따라 전체 원자 수는 MOF의 기저 구조에 따라 최소 2,856개 부터 최대 4,036개까지로 구성되었다.
시뮬레이션 구동을 위한 초기 구조의 최적화는 LBFGS 알고리즘(fmax = 0.005 eV/Å, 최대 1,000 steps)을 통해 수행되었으며, 이후 랑주뱅 써모스탯(Langevin Thermostat)을 적용하였다. 또한 원자 수(N), 부피(V), 온도(T)를 일정하게 유지하는 NVT 앙상블에서 300 K 조건으로 시뮬레이션이 진행되었으며, 이를 통해 MOF 내부에서의 이산화탄소 확산 거동을 안정적으로 모사하였다. 총 시뮬레이션 시간은 500 ps로 설정하였고, 초기 속도는 맥스웰-볼츠만 분포(Maxwell-Boltzmann Distribution)에 따라 무작위로 부여하였다. 원자 좌표는 1.0 ps 간격으로 기록하였으며, 이산화탄소의 확산계수는 평균제곱변위(Mean Square Displacement, MSD) 곡선의 200-500 ps 구간 선형 영역을 기반으로 도출하였다. 이와 같은 시뮬레이션 환경에서, NNP를 적용하여 MD 시뮬레이션을 수행하였다.
2.2 NNP 기반 MD 시뮬레이션의 도입
DFT은 양자역학을 기반으로 원자, 분자, 고체와 같은 전자 시스템의 바닥 상태(Ground State) 특성을 계산하는 방법이며, 전자 밀도를 기본 변수로 사용하여 양자 시스템의 상태를 기술한다. 그러나 계산 비용이 매우 크며 계산 자원에 따라 달라지지만 최대 500 개의 원자에 대한 정적 계산에 국한된다[
17].
MD 시뮬레이션은 원자와 분자의 동역학적 거동을 전산모사하여 시간에 따른 시스템의 구조적 및 에너지적 변화를 추적하는 방법론이다. 이 과정에서 정의된 힘장으로부터 원자 간 상호작용을 계산하고, 뉴턴 운동 방정식을 적용하여 원자 수준의 궤적을 생성한다. 최신 알고리즘과 계산 자원을 활용하면 최대 약 10
6개의 원자와 10
4 ps 규모까지 모사가 가능하다[
18]. 다만 계산 정확도는 적용된 힘장에 크게 의존하며, 새로운 화학적 환경에서의 상호작용을 신뢰성 있게 재현하기 어렵고, 화학 반응성, 결합 형성 및 파괴를 직접적으로 기술하기에는 근본적인 제약이 존재한다[
19,
20].
한편, 제1원리 분자동역학(Ab Initio Molecular Dynamics, AIMD) 시뮬레이션은 전자 구조 계산을 통해 원자 간 상호작용을 양자역학적 정확도로 도출하고, 이를 바탕으로 시간에 따른 분자의 동역학적 거동을 모사하는 접근법이다. AIMD 시뮬레이 션은 파동함수 혹은 전자 밀도에 기초하여 원자 수준의 상호작용과 에너지 변화를 직접적으로 기술할 수 있다는 장점이 있기 때문에 MOF 내 입자 거동 분석에 주로 활용된다. 그러나 계산 비용이 매우 높아 일반적으로 약 300개 이하 원자와 50 ps 이내의 시간 규모로 제한된다. 따라서 대규모 시스템이나 장시간 스케일에서의 평형 거동 모사에는 적합하지 않다[
21].
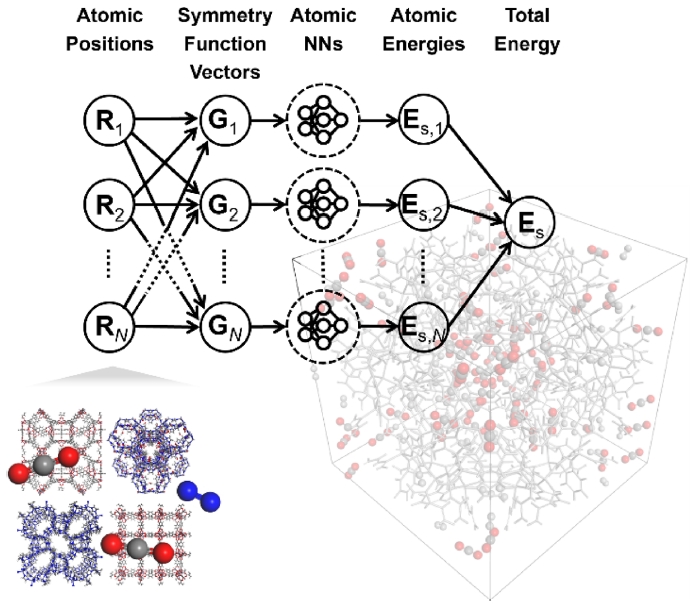
본 연구에서는 MOF 내 이산화탄소 분자의 거동을 모사하고 정량적으로 평가하기 위해 양자역학 계산과 분자 동역학을 동시에 반영할 수 있는 NNP를 도입하였다. NNP는 DFT 수준의 정확도를 유지하면서도 다양한 금속 원소와 산화물에 대한 높은 범용성을 제공한다. 이를 통해 6,000개 이상의 원자로 구성된 중 규모 이상의 시스템에 대한 분자동역학 시뮬레이션이 가능하다[
22]. 고차원 신경망 구조는 각 원자 주변의 국소 화학적 환경을 반영하여 원자 단위 에너지(E
i)를 계산하고, 전체 시스템의 총 에너지(E)는 식(1)과 같이 모든 원자 에너지의 합으로 표현된다.
NNP 기반 MD 시뮬레이션은 분자 내부 전하 분포 변화까지 반영할 수 있어 화학 반응성을 직접적으로 기술할 수 있으며, 최대 10
4개 원자로 구성된 시스템에서 수십-수백 ps 규모의 계산을 수행할 수 있다[
23]. 특히 MOF와 같은 복잡계에서 금속–산화물 간 상호작용을 정밀하게 기술할 수 있어, 기존 접근법으로는 정밀하게 평가하기 어려웠던 이산화탄소 확산 거동을 정량적으로 규명하는 데 활용될 수 있다[
24].
3. 결과 및 고찰
3.1 MOF 내 이산화탄소 입자 거동 정량평가
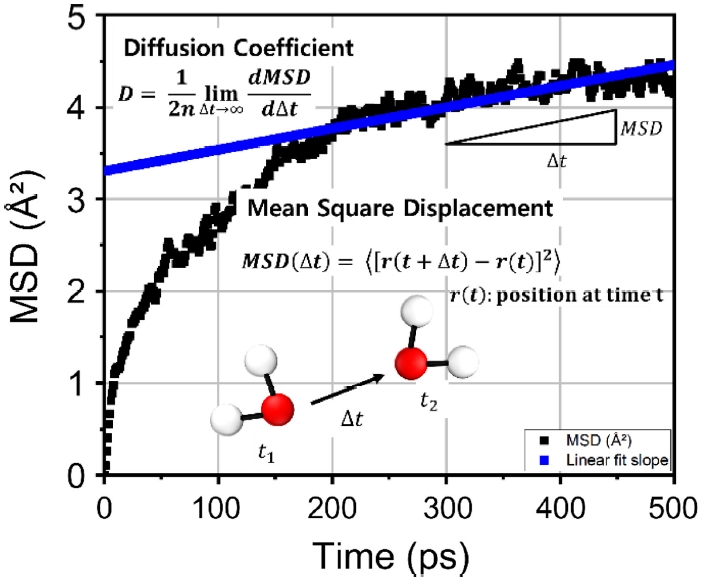
입자의 거동을 분석하는 지표로는 결합 에너지, 전하 분포, 시간에 따른 분포 변화, 그리고 온도 의존적 활성 정도 등이 고려될 수 있다. 본 연구에서는 MOF의 금속과 원자 배열에 따른 이산화탄소의 내부 거동을 정량적으로 규명하는 것이 주요 목적이므로, MSD로부터 도출할 수 있는 확산계수를 평가 지표로 설정하였다. MD 시뮬레이션은 시간에 따른 원자 및 분자의 위치 정보를 직접적으로 제공하므로, MSD는 확산 특성을 분석하는 데 가장 보편적이고 신뢰성 있는 방법론으로 활용된다. MSD는 시간 간격 Δt동안 입자의 위치 변화 제곱 평균으로 정의되며, 식(2)와 같이 표현된다.
여기서 r(t)는 시간 t에서의 입자 위치 벡터이며 r(t + Δt)는 시간 간격 Δt 이후의 위치를 의미한다. 확산계수(D)는 MSD의 시간에 대한 장시간 선형 기울기에서 도출되며, 차원 수 n을 고려하여 식(3)과 같이 정의된다. 본 연구에서는 3차원 확산을 가정하여 n = 3을 적용하였다.
확산계수는 단순한 분자 이동 거리를 넘어, 시스템 내에서 분자가 장시간에 걸쳐 얼마나 분산되는지를 정량적으로 반영하는 지표로, 개별 분자의 궤적 분석을 통해 확산 메커니즘을 규명할 수 있다. MOF의 벌크 특성을 평가하기 위해 단위셀을 기반으로 시뮬레이션을 수행하였으며, 이를 통해 각 구조에서의 이산화탄소 확산도를 비교·분석하였다. 초기 조건은 무작위로 배치된 이산화탄소 분자 배열로 설정하였고, 시스템에서 원자 수(
N), 체적(
V), 온도(
T)를 고정한
NVT 앙상블에서 시뮬레이션을 진행하였다. 시스템의 크기 및 구성 원자의 종류에 따라 평형 도달 시간은 다소 차이가 있으나, 약 200 ps 에서 시스템이 평형 상태에 도달했다고 판단하였다. 따라서 0-200 ps 비평형 구간은 분석에서 제외하였으며, 평형에 도달한 이후인 200-500 ps 구간의 데이터를 이용하였다. 해당 구간에서 MSD의 시간에 따른 선형 기울기를 도출하여 확산계수를 계산하였다.
Fig. 4는 Cd
3BTB
2에서 계산된 이산화탄소의 MSD 곡선과 기울기를 나타낸 것이다. 이를 통해 Cd
3BTB
2의 확산계수는 1.79 × 10
-19 cm
2/s로 도출되었다. 또한 가장 성능이 우수했던 ZIF-3의 확산계수는 7.63 × 10
-19 cm
2/s이다. 다만, 두 물질의 결과를 하나의 그림에 동시에 제시할 경우 그래프가 복잡해져 시각적 명확성이 저하될 우려가 있어,
Fig. 4에는 Cd
3BTB
2의 결과만 제시하였다.
MOF 내 이산화탄소 확산계수 평가를 위한 시스템을 구축하였다. 각 MOF의 결정 구조를 모델링한 뒤 500 ps 동안 시뮬레이션을 수행하여 확산계수를 도출하였다.
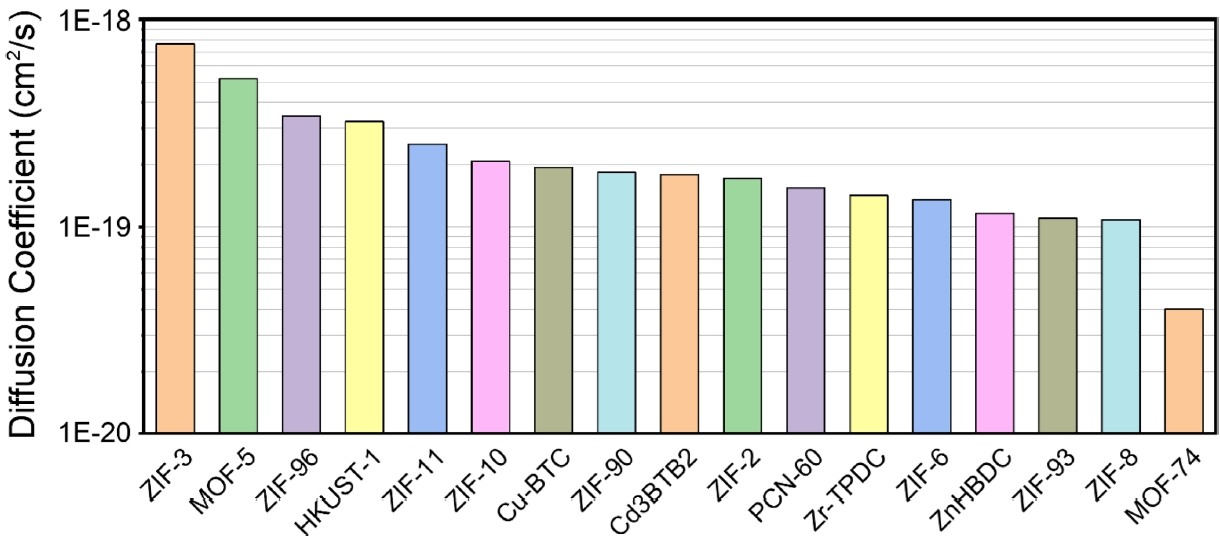
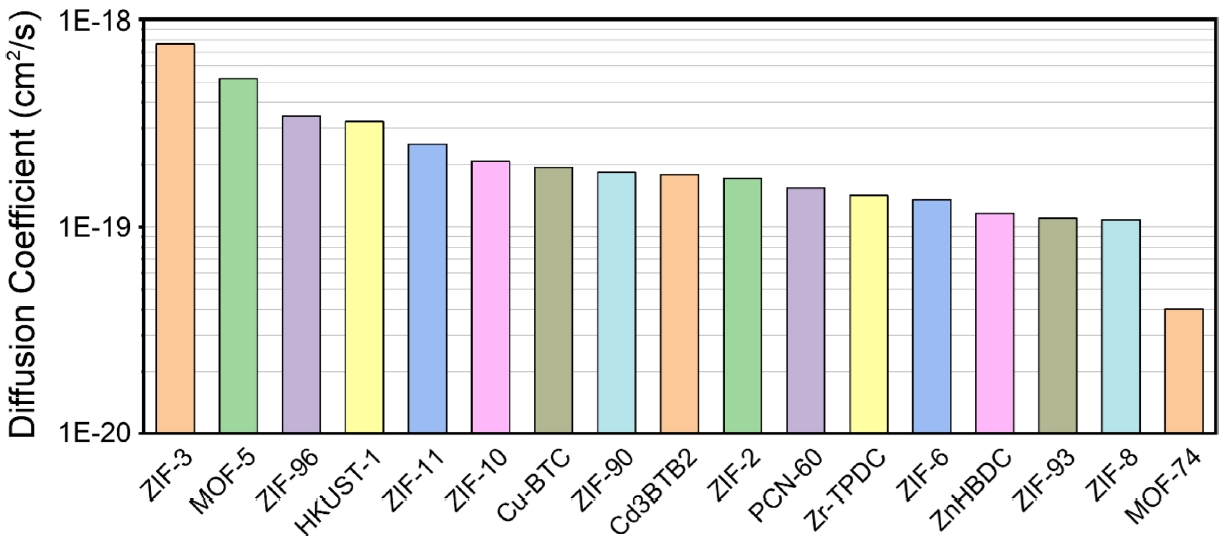
Fig. 5는 MOF 17종에서 계산된 이산화탄소 확산계수 결과를 나타낸 그래프이다.
그 결과, MOF의 유형에 따라 이산화탄소의 확산 성능에는 차이가 확인되었다. ZIF-3는 7.63 × 10
-19 cm
2/s로 가장 높은 확산계수를 보였으며, MOF-74는 4.01 × 10
-20 cm
2/s로 가장 낮은 값을 보여 두 물질 간 약 19배의 차이가 존재하였다. 이러한 차이는 ZIF-3의 상대적으로 개방적이고 유연한 기공 구조가 확산을 촉진한 반면, MOF-74는 일차원적 채널 구조로 분자의 이동이 제한된 결과로 해석할 수 있다. 따라서 MOF의 구조적 차이에 따라 이산화탄소 확산 특성이 달라질 수 있음을 보여준다. ZIF-3, MOF-5, ZIF-96, ZIF-93의 CO
2 확산계수에 대한 실험치는 보고되어 있지 않으나, 문헌에서 ZIF-8 ((1.4-2.2) × 10
-6 cm
2/s) [
25]이 MOF-74 ((3-8) × 10
-7 cm
2/s) [
26]보다 빠른 확산을 보인다는 결과가 보고되어 있다. 이러한 실험 경향은 본 연구의 시뮬레이션 결과와 일치한다.
추가적으로 MOF 내 이산화탄소의 확산 메커니즘을 보다 정밀하게 규명하기 위해서는 금속 이온과 이산화탄소 분자 간의 상호작용 에너지를 정량적으로 평가할 필요가 있다. 이를 위해 DFT 기반의 결합 에너지 계산을 수행하거나, 흡착 위치별 에너지 분포를 비교·분석하는 접근법을 적용할 수 있다. 이러한 정량적 분석을 통해 MOF별 금속–기체 상호작용 강도를 평가함으로써, 확산 거동 차이의 근본적 원인을 명확히 규명할 수 있다. 또한, 금속 이온을 기준으로 한 이산화탄소의 공간적 분포를 확인하기 위해 동경분포함수(Radial Distribution Function) 분석을 수행하면, 기공 내부에서의 분자 배치, 선호 흡착 거리, 및 상호작용 빈도를 효과적으로 파악할 수 있다. 이러한 분석을 병행함으로써 MOF 구조적 특성과 확산 특성 간의 상관관계를 정량적으로 해석할 수 있을 것으로 기대된다.
4. 결론
대기 중 이산화탄소 농도의 증가는 탄소 포집·저장(CCS) 기술의 필요성을 더욱 부각시키고 있으며, 이 중에서도 분리·회수 단계의 포집 기술은 핵심적이다. MOF는 높은 흡착 용량과 구조적 설계 가능성을 바탕으로 유망한 고상 흡수제로 주목받고 있다. 한편, 기존 계산화학적 방법론만으로는 금속과 산화물 간 전자적 상호작용과 입자 거동을 동시에 정밀하게 모사하기 어려웠다. 본 연구에서는 이를 극복하기 위해 NNP 기반 MD 시뮬레이션을 도입하였고, 17종 MOF의 이산화탄소 확산계수를 도출하였다. 그 결과, ZIF-3는 가장 높은 확산계수를, MOF-74는 가장 낮은 값을 나타내어 두 물질 사이 약 19배 차이를 확인하였다. 이는 MOF의 구조적·화학적 환경이 확산 메커니즘에 일정 부분 기여함을 보이며, NNP 기반 접근을 통해 정량적 평가가 가능한 것을 나타내었다. 또한 단순화된 모델에서도 의미 있는 비교가 가능함을 확인하였으며, 본 연구는 MOF 기반 이산화탄소 포집 소재의 설계 과정에서 확산 특성을 주요 영향 인자로 고려한다면, 중요 지표로 활용될 수 있다. 이를 바탕으로 향후 실험 검증 및 조건 확장을 통해 보다 정교한 평가가 가능할 것으로 기대된다.
FOOTNOTES
-
ACKNOWLEDGEMENT
본 과제(결과물)는 2025년도 교육부 및 광주광역시의 재원으로 광주RISE센터의 지원을 받아 수행된 지역혁신중심 대학지원체계(RISE) 사업의 결과임(No. 2025-RISE-05-011). 본 연구성과는 2025년도 정부(교육부)의 재원으로 한국연구재단의 지원을 받아 수행된 기초연구사업임(No. RS-2025-25430676).
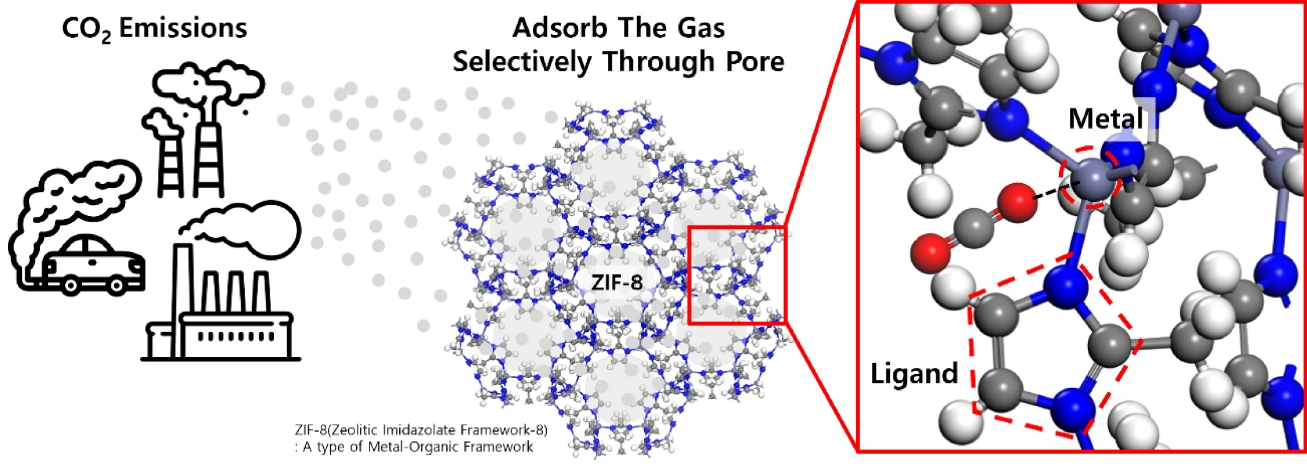
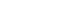
Fig. 1Schematic illustration of CO2 capture in a metal-organic framework structure.
Fig. 2Metal–organic frameworks (MOFs) evaluated in this study
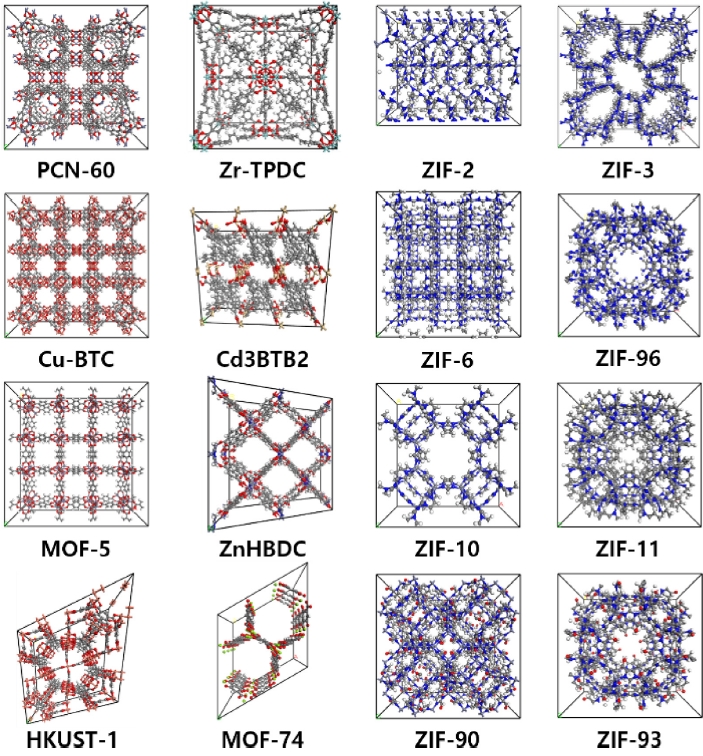
Fig. 3Schematic of neural network potential (NNP) molecular dynamics methodology
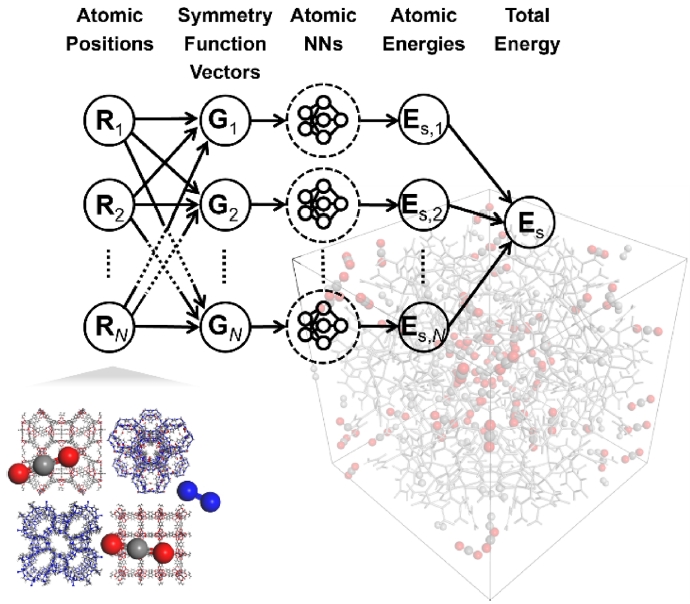
Fig. 4Evaluation of CO2 diffusion coefficient in metal–organic framework (MOF) via mean square displacement (MSD) analysis
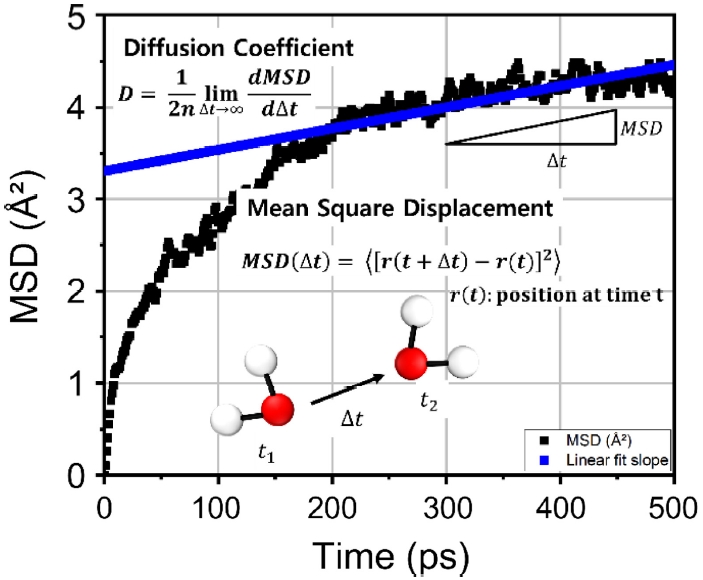
Fig. 5Comparison of CO2 diffusion coefficients in metal–organic frameworks
REFERENCES
- 1. Kabir, M., Habiba, U. E., Khan, W., Shah, A., Rahim, S., De los Rios-Escalante, P. R., Farooqi, Z.-U.-R., Ali, L., Shafiq, M., (2023), Climate change due to increasing concentration of carbon dioxide and its impacts on environment in 21st century; a mini review, Journal of King Saud University-Science, 35(5), 102693.
- 2. Mimura, N., (2013), Sea-level rise caused by climate change and its implications for society, Proceedings of the Japan Academy, Series B, 89(7), 281-301.
- 3. Solomon, S., Plattner, G.-K., Knutti, R., Friedlingstein, P., (2009), Irreversible climate change due to carbon dioxide emissions, Proceedings of the National Academy of Sciences, 106(6), 1704-1709.
- 4. Hanson, E., Nwakile, C., Hammed, V. O., (2024), Carbon capture, utilization, and storage (CCUS) technologies: Evaluating the effectiveness of advanced CCUS solutions for reducing Co2 emissions, Results in Surfaces and Interfaces, 100381.
- 5. Wilberforce, T., Olabi, A., Sayed, E. T., Elsaid, K., Abdelkareem, M. A., (2021), Progress in carbon capture technologies, Science of The Total Environment, 761, 143203.
- 6. Songolzadeh, M., Soleimani, M., Takht Ravanchi, M., Songolzadeh, R., (2014), Carbon dioxide separation from flue gases: A technological review emphasizing reduction in greenhouse gas emissions, The Scientific World Journal, 2014(1), 828131.
- 7. Li, J.-R., Kuppler, R. J., Zhou, H.-C., (2009), Selective gas adsorption and separation in metal–organic frameworks, Chemical Society Reviews, 38(5), 1477-1504.
- 8. Kitagawa, S., (2014), Metal–organic frameworks (MOFS), Chemical Society Reviews, 43(16), 5415-5418.
- 9. Ding, M., Flaig, R. W., Jiang, H.-L., Yaghi, O. M., (2019), Carbon capture and conversion using metal–organic frameworks and mof-based materials, Chemical Society Reviews, 48(10), 2783-2828.
- 10. Kong, X., Scott, E., Ding, W., Mason, J. A., Long, J. R., Reimer, J. A., (2012), Co2 dynamics in a metal–organic framework with open metal sites, Journal of the American Chemical Society, 134(35), 14341-14344.
- 11. Pettinari, C., Marchetti, F., Mosca, N., Tosi, G., Drozdov, A., (2017), Application of metal− organic frameworks, Polymer International, 66(6), 731-744.
- 12. Queen, W. L., Hudson, M. R., Bloch, E. D., Mason, J. A., Gonzalez, M. I., Lee, J. S., Gygi, D., Howe, J. D., Lee, K., Darwish, T. A., (2014), Comprehensive study of carbon dioxide adsorption in the metal–organic frameworks M2 (dobdc)(M= Mg, Mn, Fe, Co, Ni, Cu, Zn), Chemical Science, 5(12), 4569-4581.
- 13. Li, P., Merz Jr, K. M., (2017), Metal ion modeling using classical mechanics, Chemical Reviews, 117(3), 1564-1686.
- 14. Boyd, P. G., Moosavi, S. M., Witman, M., Smit, B., (2017), Force-field prediction of materials properties in metal-organic frameworks, The Journal of Physical Chemistry Letters, 8(2), 357-363.
- 15. Lin, R.-B., Xiang, S., Zhou, W., Chen, B., (2020), Microporous metal-organic framework materials for gas separation, Chem, 6(2), 337-363.
- 16. Takamoto, S., Shinagawa, C., Motoki, D., Nakago, K., Li, W., Kurata, I., Watanabe, T., Yayama, Y., Iriguchi, H., Asano, Y., (2022), Towards universal neural network potential for material discovery applicable to arbitrary combination of 45 elements, Nature Communications, 13(1), 2991.
- 17. Fiedler, L., Modine, N. A., Schmerler, S., Vogel, D. J., Popoola, G. A., Thompson, A. P., Rajamanickam, S., Cangi, A., (2023), Predicting electronic structures at any length scale with machine learning, npj Computational Materials, 9(1), 115.
- 18. Thompson, A. P., Aktulga, H. M., Berger, R., Bolintineanu, D. S., Brown, W. M., Crozier, P. S., In't Veld, P. J., Kohlmeyer, A., Moore, S. G., Nguyen, T. D., (2022), Lammps-a flexible simulation tool for particle-based materials modeling at the atomic, meso, and continuum scales, Computer Physics Communications, 271, 108171.
- 19. Senn, H. M., Thiel, W., (2009), Qm/mm methods for biomolecular systems, Angewandte Chemie International Edition, 48(7), 1198-1229.
- 20. Chmiela, S., Sauceda, H. E., Müller, K.-R., Tkatchenko, A., (2018), Towards exact molecular dynamics simulations with machine-learned force fields, Nature Communications, 9(1), 3887.
- 21. Zhang, Y., Maginn, E. J., (2012), A simple AIMD approach to derive atomic charges for condensed phase simulation of ionic liquids, The Journal of Physical Chemistry B, 116(33), 10036-10048.
- 22. Matsumura, N., Yoshimoto, Y., Yamazaki, T., Amano, T., Noda, T., Ebata, N., Kasano, T., Sakai, Y., (2025), Generator of neural network potential for molecular dynamics: Constructing robust and accurate potentials with active learning for nanosecond-scale simulations, Journal of Chemical Theory and Computation, 21(8), 3832-3846.
- 23. Shaidu, Y., Smith, A., Taw, E., Neaton, J. B., (2023), Carbon capture phenomena in metal-organic frameworks with neural network potentials, PRX Energy, 2(2), 023005.
- 24. Zheng, B., Oliveira, F. L., Neumann Barros Ferreira, R., Steiner, M., Hamann, H., Gu, G. X., Luan, B., (2023), Quantum informed machine-learning potentials for molecular dynamics simulations of Co2’s chemisorption and diffusion in Mg-MOF-74, ACS Nano, 17(6), 5579-5587.
- 25. Pusch, A.-K., Splith, T., Moschkowitz, L., Karmakar, S., Biniwale, R., Sant, M., Suffritti, G. B., Demontis, P., Cravillon, J., Pantatosaki, E., (2012), Nmr studies of carbon dioxide and methane self-diffusion in zif-8 at elevated gas pressures, Adsorption, 18(5), 359-366.
- 26. Forse, A. C., Colwell, K. A., Gonzalez, M. I., Benders, S., Torres-Gavosto, R. M., Blümich, B., Reimer, J. A., Long, J. R., (2020), Influence of pore size on carbon dioxide diffusion in two isoreticular metal–organic frameworks, Chemistry of Materials, 32(8), 3570-3576.
Biography
- JeongMin Shin
B.Sc. candidate in the School of Mechanical Engineering, Chonnam National University. Her research interest is multiscale computational modeling for energy materials.
- Sangbaek Park
Professor in the Department of New Materials Engineering, Chungnam National University. His research interest is advanced materials and devices for energy storage an conversion.
- JinHyeok Cha
Professor in the Scool of Mechanical Engineering, Chonnam National University. His research interest is machine learning based multiscale modeling for advanced materials